Research interest
One goal of my research is to understand how biological species
and their genomes evolve and how this is determined by their current
genome composition, structure, interactions between genes within genomes and individuals
or species within ecological habitats.
A second and related goal of my research is to contribute to our
understanding of inter and intra specific microbe-microbe interactions and their mechanisms.
I approach these questions via developing and applying
novel statistical and computational protocols to genomics data.
A lot of my methods combine evolutionary, systems biology, phylogenetics and genomics and
bridge the gaps between the disciplines.
Predicting and understanding microbe-microbe interactions
Currently microbe-microbe interactions are inferred from concerted changes in abundance of lineage specific 16S rRNA in ecological samples.
This approach, while fruitful, does not provide a handle on molecular mechanisms of the interactions.
Genomics might hold a key to solving the problem.
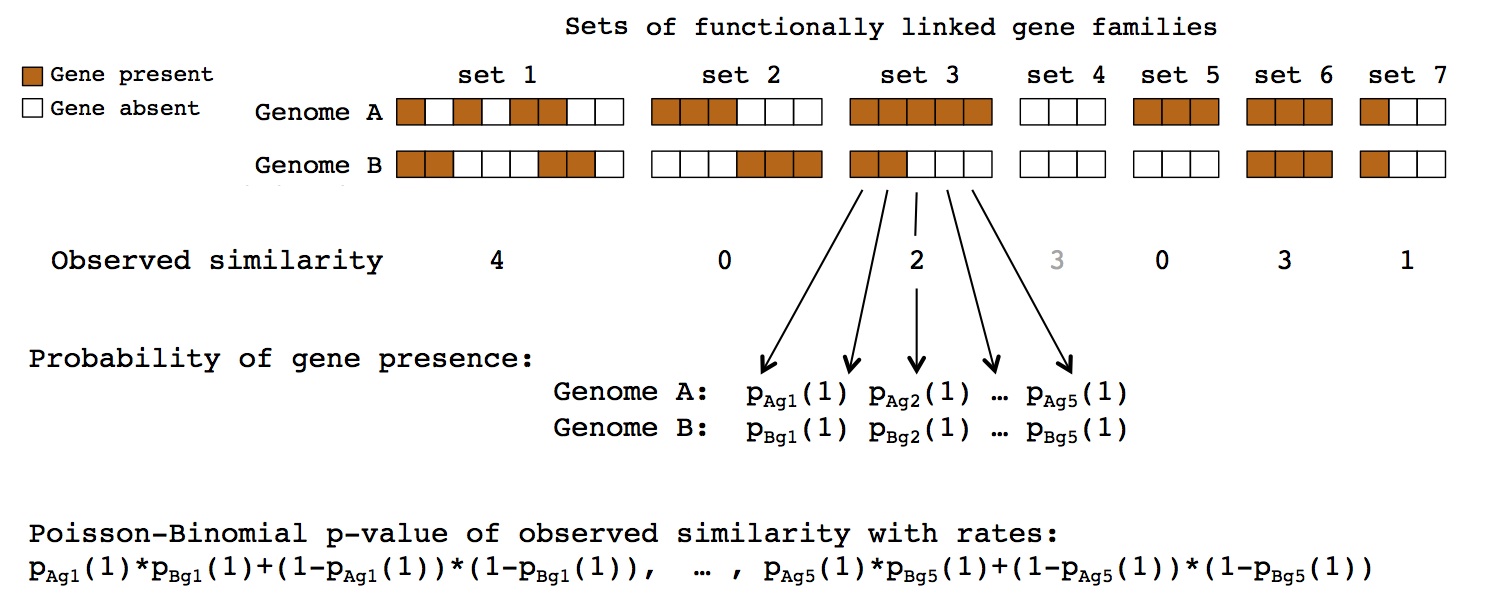
To this end, I developed two new genomics-based indices to evaluate propensities of microbial species to interact with each other.
One index is based upon analysis of the protein-protein functional association network built jointly for two bacteria genomes.
Another one quantifies functional similarity between the genomes and also based on predictions of functional association between proteins.
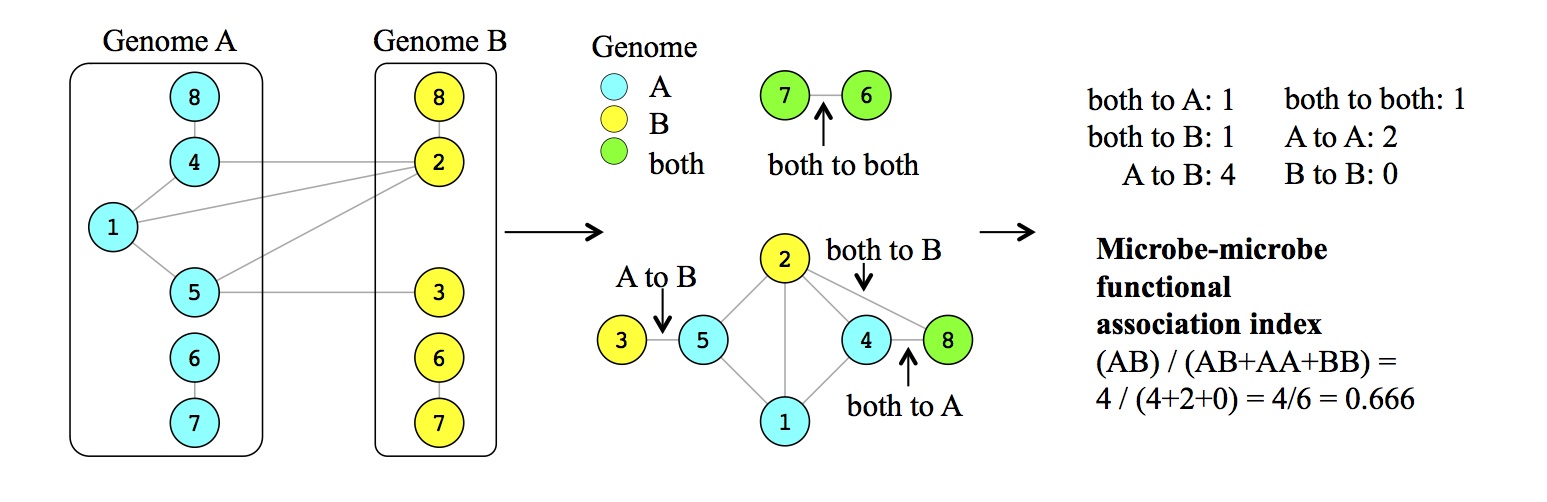
I applied new metrics to hundreds of microbial genomes from human microbiota and divergent ecological habitats.
Using 16S rRNA based predictions of microbial interactions I showed that new indices explain about 5% of variance in microbial co-occurrence,
which constitutes 1.5 time improvement on predictive ability of previously existing methods
(
Kamneva. PLoS Comp Biol. 2017).
Another outstanding knowledge gap in microbial ecology is a lack of methods for identifying genes or pathways potentially facilitating
microbe-microbe interactions between known partners. To address this issue, I designed new method to detect genes, or sets of functionally linked genes,
which evolve none-independently in terms of their gain and loss in genomes of interacting partners.
This pattern is expected for genes facilitating interaction between the species on molecular level.
I applied this new method to systematically predict potential genes or gene sets mediating interactions between
Comamonadaceae bacterium CR and
Chlorobium chlorochromatii CaD3, members of phototrophic consortium Chlorochromatium aggregatum,
representing one of the classic examples of microbial symbiosis (
Kamneva. In preparation).
Statistical phylogenetics
I also have interest in statistical phylogenetics, in particular, in detecting and understanding horizontal processes in species evolution
resulting in gene tree species tree discordance (Kamneva & Ward. In Methods in Microbiology. 2014).
I am involved in empirical data analysis project concerned with detecting hybridizations in
Fragaria species (Kamneva et al. In revision).
I also study available methods for detecting hybridization events using simulated data
and develop new analysis protocols (Kamneva & Rosenberg. Evol Bioinf. 2017).
Microbial evolutionary, comparative and functional genomics
This line of work combines statistical phylogenetics, functional and bioinformatic analysis.
I study how changes in genome composition and protein-coding genes are correlated with acquisition and loss of cellular traits in bacteria using cellular structure of bacteria from
Planctomycetes-Verrcomicrobia-Chlamydiae (PVC) super-phylum as model group (Kamneva et al. In New Models. 2013).
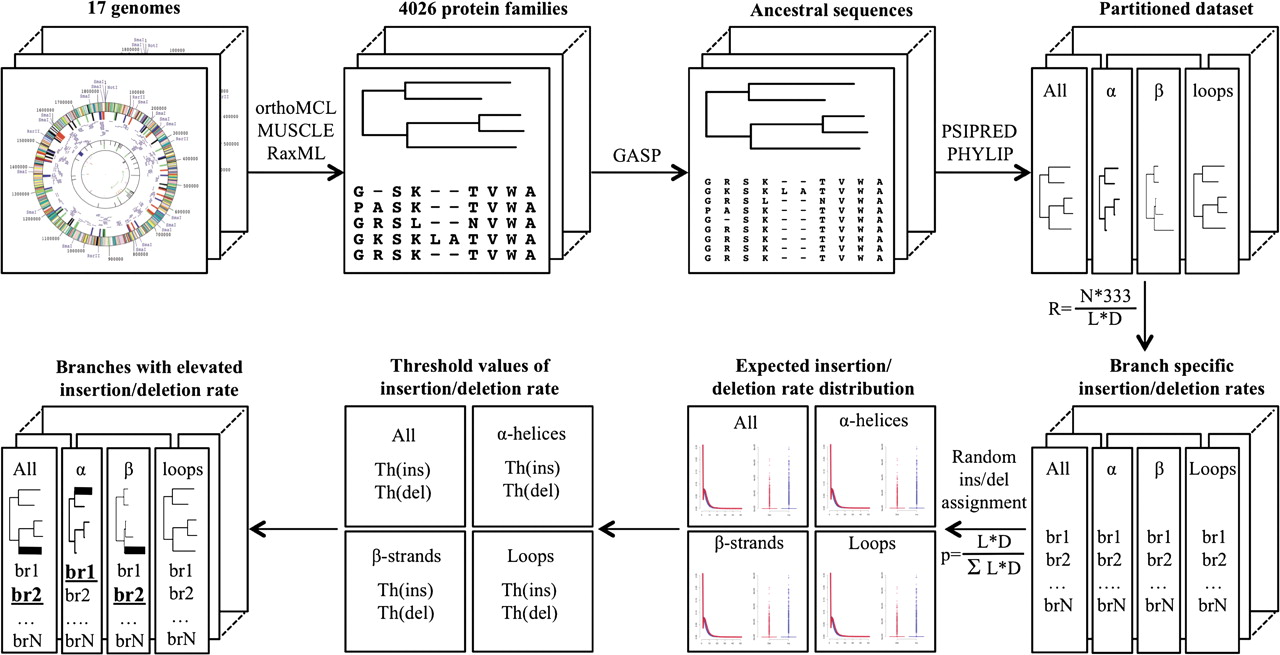
I developed a novel computational framework for quantifying selective pressure on indel substitutions within protein-coding genes
which is applicable to distantly related organisms.
Using this new method identified a number of gene families and several biochemical pathways where indels occur more frequently
than expected by chance (
Kamneva et al. GBE. 2010).
I also characterized patterns of genome content evolution in these bacteria using gene-tree species-tree reconciliation technique followed by
Bayesian mixture-modeling of evolutionary events rates (Kamneva et al. GBE. 2012)
showing that a large number of genes were acquired on various PVC lineages from phylum Acidobacteria
and from phylum Bacteroidetes on the lineage leading to Akkermansia muciniphila, an intestinal human commensal.
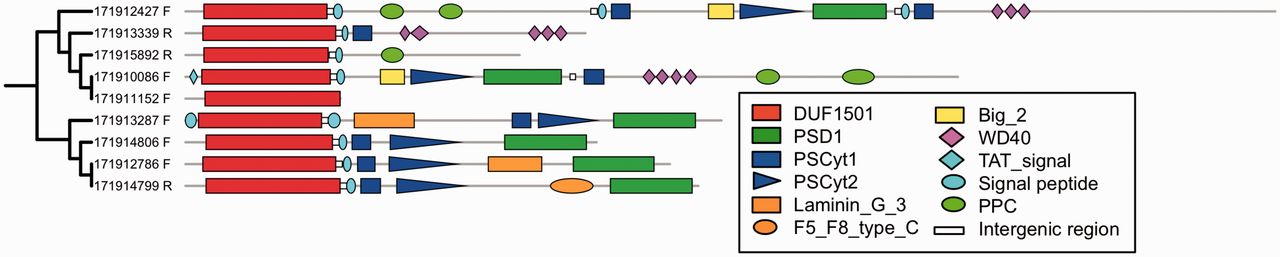
Systems biology inspired part of this study has allowed me to identify a highly conserved genetic module which includes two to four genes
statistically associated with the complex PVC cell plan.
Functional sequence analysis has allowed me to identify signal peptide sequences over-represented in protein families preferentially present in
membrane-bearing PCV bacteria suggesting Sec-mediated mechanism of their targeting (Kamneva et al. PLoS ONE. 2015).
My work also includes molecular evolutionary studies on snakes (Kvon et al. Cell. 2016), Firmicutes (Volkov et al. JBC. 2010) and Verrucomicrobia
(Sait et al. Front Microbiol. 2011)
as well as bioinformatic analysis on Shigella flexneri genome (Hensley et al. Arch Microbiol. 2011).
Publications
Follow link for full Google scholar profile
OK Kamneva.
Understanding mechanisms of microbe-microbe interactions using patterns of genome content changes. In preparation.
OK Kamneva, J Syring, A Liston, NA Rosenberg.
Evaluating allopolyploid origin of Fragaria species using 257 nuclear loci. In revision for BMC Evolutionary Biology.
OK Kamneva, NA Rosenberg (2017)
Simulation-based evaluation of species network reconstruction methods in presence of incomplete linage sorting.
Evolutionary Bioinformatics. 13. [PDF]
OK Kamneva (2017)
Genome content of microbes predicts their co-occurrence in the environment.
PLoS Computational Biology. 13 (2): e1005366. [PDF]
EZ Kvon, OK Kamneva, US Melo, et al. (2016)
Progressive loss of function in a limb enhancer during snake evolution.
Cell. 167 (3): 633-642. [PDF]
OK Kamneva, S Poudel, NL Ward (2015)
Proteins related to the Type I secretion system are associated with secondary SecA_DEAD domain proteins in some species of Planctomycetes, Verrucomicrobia, Proteobacteria, Nitrospirae and Chlorobi.
PLoS ONE. 6: e0129066. [PDF]
OK Kamneva, NL Ward (2014)
Reconciliation approaches to determining HGT, duplications and losses in gene trees.
Chapter in "Methods in Microbiology", 41: 183-199. [PDF]
OK Kamneva, DH Haft, SJ Knight, DA Liberles, NL Ward (2013)
Genomics and bioinformatics of the PVC superphylum.
Chapter in "New Models for Cell Structure, Origins and Biology: Planctomycetes". 165-193. Springer. [PDF].
OK Kamneva, SJ Knight, DA Liberles, NL Ward (2012)
Analysis of genome content evolution in PVC super-phylum: assessment of candidate genes associated with cellular compartmentalization and host-dependent life-style in bacteria.
Genome Biology and Evolution. 4: 375-390. [PDF]
M Sait, OK Kamneva, DS Fay, NV Kirienko, J Polek, MM Shirasu-Hiza, NL Ward (2011) Genomic and experimental evidence suggests that Verrucomicrobium spinosum interacts with eukaryotes. Frontiers in Microbiology. 2: 211. [PDF]
CT Hensley, OK Kamneva, KM Levy, SK Labahn, LA Africa, HJ Wing (2011) Two promoters and two translation start sites control the expression of the Shigella flexneri outer membrane protease IcsP. Archives of Microbiology. 193: 263-274. [PDF]
OK Kamneva, DA Liberles, NL Ward (2010) Genome-wide influence of indel substitutions on evolution of bacteria of the PVC super-phylum, revealed using a novel computational method. Genome Biology and Evolution. 2: 870-886. [PDF]
A Volkov, A Liavonchanka, O Kamneva, T Fiedler, C Goebel, B Kreikemeyer, I Feussner (2010) Myosin cross-reactive antigen of Streptococcus pyogenes M49 encodes a fatty acid double bond hydratase that plays a role in oleic acid detoxification and bacterial virulence. Journal of Biological Chemistry. 285: 10353-10361. [PDF]
AV Katokhin, VM Ephimov, MSh Badratinov, OK Kamneva, VA Mordvinov (2006) Multidimensional analysis and functional assignment of DNA-microarray transcription profiles of genes involved in adipogenesis. Biophysics. 51: 100-109. [PDF]